Why Medical Device Classification Matters
 When it comes to commercializing medical devices in the United States, manufacturers must manage many moving parts—from establishing quality management processes and effective design controls to preparing the necessary documentation and clinical evidence for FDA approval.
When it comes to commercializing medical devices in the United States, manufacturers must manage many moving parts—from establishing quality management processes and effective design controls to preparing the necessary documentation and clinical evidence for FDA approval.
The first step in obtaining FDA approval is classifying your medical device. This is a critical step, as it determines the type of premarket submissions and applications that will be required by the FDA and creates a roadmap for compliance throughout the product lifecycle. More importantly, the device classification establishes the proper controls to ensure patient safety.
Here, we outline the key steps for classifying your medical device and reveal how a cloud-based enterprise quality management software system (eQMS) can help streamline FDA compliance.
Differences Between Class I, II, and III Devices
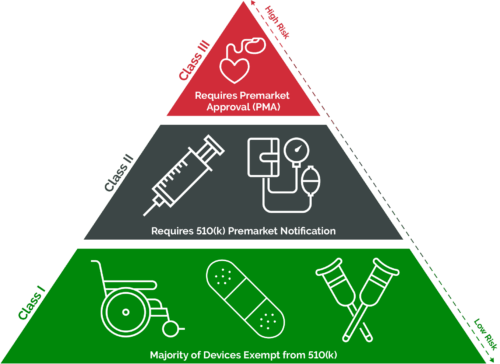
The FDA categorizes medical devices as either Class I, Class II, or Class III. This three-tier classification is based on the medical device’s intended use, indications for use, and the risk that it poses. Different regulatory controls (i.e., general, special, or premarket approval) are assigned to each class to ensure the safety and effectiveness of the device. The amount of regulatory control increases as devices progress from Class I to Class III.
Class I devices
Almost 50% of medical devices in the U.S. market fall into the Class I category. These devices pose the lowest risk to patients and/or users and are only subject to general controls such as adverse event reporting and device tracking. Most devices in this category are exempt from 510(k) premarket notification and FDA clearance. Examples of Class I devices include bandages, wheelchairs, and manual stethoscopes.
Class II devices
Class II devices pose a moderate to high risk to patients and/or users. These devices are subject to both general controls and special controls, such as post-market surveillance and special labeling requirements. Unlike Class I devices, most Class II devices require a 510(k) submission to obtain FDA clearance. Today, over 40% of medical devices fall into the Class II category. Examples include catheters, syringes, and pregnancy test kits.
Class III devices
Class III devices pose the highest risk to patients and/or users and are subject to both general controls and premarket approval (PMA). As part of the PMA application process, companies must submit extensive scientific evidence in the form of clinical trial data to demonstrate the safety and effectiveness of the device. Class III devices are typically implantable and designed to help sustain or support human life. Only 10% of medical devices fit into this category. Examples include defibrillators, pacemakers, and breast implants.
3 Steps to Classify Your Medical Device
Misclassification of your medical device can be a costly mistake, due to unnecessary time and money spent on data collection, document preparation, and application fees. To ensure that your device is classified correctly, follow these three steps:
1. Determine the intended use and indications for use of your medical device.
Prior to classifying your device, you should formulate clear statements regarding its intended use and indications of use.
- Intended use defines the general purpose or function of the medical device.
- Indications for use describe the disease or condition the medical device will diagnose, prevent, treat, or cure. They also specify the target patient population.
2. Select the FDA medical specialty and associated regulation(s) that align with your device.
The FDA groups medical devices into 16 different panels or specialties as part of its classification system. These specialties are listed in CFR Title 21: Parts 862 to 892.

Source: CFR – Code of Federal Regulations Title 21
Carefully review the medical specialties and select the one that is the best fit for your device. When you click on a specialty of interest, it will display a list of corresponding regulations.
Example: Below is a partial list of the regulations that correspond to the 870 Cardiovascular devices specialty:

Source: CFR – Code of Federal Regulations Title 21 Part 870
Carefully review the regulations and select the ones that best align with your device based on its intended use and indications for use.
Example: Below is the information that displays for the 870.5325 Defibrillator tester regulation:

Source: CFR – Code of Federal Regulations Title 21 Part 870
3. Find the FDA product codes that are applicable to your device.
Once you’ve selected the applicable regulations, you will need to find the product codes that pertain to your device by conducting a search in the FDA Product Classification Database.
Source: FDA Product Classification
Enter the regulation numbers that were selected in Step 2 and click the search button. You will get a detailed listing for the regulation that includes a product code and corresponding device class. Additional information, including the FDA submission type, is also highlighted in the listing.
Example: Product code and device class listing for regulation 870.5325:

Source: FDA Product Classification
You will need to perform a separate product code search for each regulation that is identified in Step 2.
This process may take some time; however, it is important to carefully review the different regulations and product codes to ensure that you choose the correct classification for your device.
Cloud QMS Solutions Simplify FDA Compliance
Once the appropriate classification is established for your device, you’ll have a clear understanding of the level of design controls, testing, and documentation needed for FDA clearance. By leveraging a cloud-based enterprise quality management system like Arena QMS, you can better manage all the product specifications, validation documentation, and clinical data that are generated for your medical device. And because this information is electronically linked to the entire product record (e.g., bill of materials, product requirements, quality processes, etc.), you can easily track and control any revisions that are made throughout the product development process to ensure you’re audit-ready and FDA compliant.
To learn more about how an enterprise quality management software system (eQMS) can help, read this best practice article.